NMR- og IR-spektroskopi som valgfrit emnePreben Albertsen, Per Bøgeholdt, Tove Eriksen og Annette Nyvad IntroduktionI dette afsnit beskrives anvendelsen af forskellige edb-programmer i forbindelse med et valgfrit forløb på højniveau (evt. mellemniveau) om spektroskopi. Hovedvægten er lagt på NMR-spektroskopi, men IR-spektroskopi indgår også i mindre omfang i forløbet. Specielt i forbindelse med identifikation af ukendte forbindelser er det en stor fordel, at man anvender begge typer spektre i processen. Det forudsættes, at eleverne i starten af dette undervisningsforløb lærer grundlæggende teori om NMR- og IR-spektroskopi. Her kan man benytte forskelligt materiale fx: — Erik Strandgaard Andersen: Spektroskopiske metoder – en elementær indføring, Gyldendal. — Materialet, som Syddansk Universitet Odense bruger til deres Efterårslaboratorium, er til fri afbenyttelse. Materialet kan hentes fra kemis hjemmeside på følgende adresse: www.ke.gymfag.dk Introduktionen kan med fordel kombineres med eller erstatte arbejde med nogle af de neden for nævnte programmer. På baggrund af denne elementære indføring i spektroskopiske metoder forventes eleverne nu at være i stand til at tolke simple spektre. I det følgende beskrives anvendelsen af forskellige programmer, der med fordel kan inddrages i forbindelse med tolkning af spektre og identifikation af ukendte stoffer. Sidst i kapitlet findes en række opgaver i tolkning af NMR- og IR-spektre. Det drejer sig om følgende tre kategorier af opgaver: — Web-baserede opgaver, hvor der ydes interaktiv hjælp til tolkningen. Opgavernes webadresse er angivet i teksten. — Opgaver, der lægger op til brug af et eller flere af de programmer, der introduceres i dette kapitel. — Opgaver, der kan løses udelukkende ved brug af Databog fysik kemi.
Hvor hentes NMR-, IR- og MS- spektre?IR og MS spektre kan hentes direkte fra: HNMR- og CNMR-spektre kan mod betaling beregnes i ACD’s Interactive lab: http://www.acdlabs.com På kemis hjemmeside www.ke.gymfag.dk kan man endvidere gratis hente beregnede H NMR-
og C NMR-spektre fra ACD, for et udvalg af de organiske forbindelser som findes i Databog
fysik kemi. NMR-spektrene er pakkede i filen work2.zip (1,5Mb). Når filen
er hentet, skal den udpakkes ved hjælp af fx WinZip, som er
sharewareprogram. I det følgende antages, at udpakningen sker til et katalog med navnet Work2.
WinZip-programmet kan hentes mange steder. Se fx henvisninger på sektornettet, adresse: Filerne er navngivet efter sidetal og nummer i Databog fysik kemi (7. udgave). Filen 104_08.hsp er således H NMR-spektret af benzen. Filen 104_08.csp er C NMR-spektret af benzen. Spektrene kan ses med programmerne HNMR-viewer eller CNMR-viewer, som omtales i næste afsnit. HNMR-viewer og CNMR-viewerDisse programmer kan vise henholdsvis H NMR- og C NMR-spektre, som er beregnet hos ACD. De beregnede spektre kan sammenlignes med eksperimentelle spektre og således fx være med til at af- eller bekræfte, om man har fremstillet det forventede stof. Eventuelle urenheder i det fremstillede stof vil ligeledes kunne påvises i spektret. Anvendelse af programmerne giver en god øvelse i fortolkning af NMR spektre. NMR-viewerne (især CNMR-vieweren) giver brugeren mulighed for at ændre på forskellige parametre, således at programmerne giver en god simulering af interaktiv spektrometerkørsel.
Installation af programmerneFilerne H-NMRv30.exe og C-NMR26.exe fra ACD kan hentes fra kemis hjemmeside. Programmerne er til Win95 eller NT. De hentede filer er installationsprogrammer. De skal kun udføres én gang. Derefter er de egentlige programmer installerede, og der er lavet genveje, som kan bruges til at starte programmerne. HNMRviewer. Fremvisning af beregnede spektre
figur 1 Spektret vises i fuld skala med kemiske skift fra –1 til 11 ppm. Ved en spektrometerfrekvens på 400 MHz svarer det til en frekvensskala for kemiske skift fra -400 til 4400 Hz .
KoblingsmønsteretSom det fremgår af spektret, er det vanskeligt at se koblingsmønsteret. Programmet giver mulighed for at zoome ind på udvalgte områder i spektret ved at trykke på højre musetast og, med venstre musetast holdt nede, trække hen over det valgte område (alternativt anvende værktøjsknappen eller Tools | Zoom In).
figur 2 Øverst ses spektret i fuld ppm skala med en markering af det valgte område. Nedenunder ses det "ind-zoomede" spektrum for det udvalgte område. Koblingsmønsteret i den valgte top ses nu tydeligt. Kemiske skift i ppm er naturligvis uafhængige af den valgte spektrometerfrekvens. I modsætning hertil er kemiske skift i MHz ligefrem proportionale med spektrometerfrekvensen. En almindelig anvendt spektrometerfrekvens er 60 MHz. Man kan vælge denne spektrometerfrekvens ved at trykke på 400 MHz på statuslinien, indtaste 60 MHz, og trykke på Recalc.
figur 3 Koblingskonstanter er uafhængige af den anvendte spektrometerfrekvens. Derfor ser man en tydeligere opsplitning af toppene, når man anvender en lavere spektrometerfrekvens.
Angivelse af dataVed at trykke på Show kan man nu få vist diverse data vedrørende spektret: Chemical Shifts, Line Intensities, Group Numbering, Integral Curve, Structure Window, Table of Chemical Shifts, Table of Coupling Constants.
figur 5 Ved at køre musen henover spektret kan man direkte se sammenhængen mellem spektrets data og strukturformlen.
OpløsningsmidletACD-spektrene er beregnet under den antagelse, at stofferne er opløst i aprotiske, upolære, ikke aromatiske opløsningsmidler (fx CCl4, CDCl3). Hvis man ønsker at sammenligne et beregnet spektrum med et eksperimentelt bestemt spektrum, som er optaget med stoffet opløst i andre typer opløsningsmidler, kan man tilpasse spektret ved at indtaste korrigerede skiftværdier i Chemical shifts tabellen og trykke på Recalculate spektrum i værktøjslinjen.
Simulering ved indtastning af tabelværdier for kemisk skift og koblingskonstanterTabelmaterialet kan hentes på kemis hjemmeside www.ke.gymfag.dk/ikt
Her ses indtastede omtrentlige værdier for ethanolmolekylet. Mere præcise data kan evt. fås fra eksperimentelle spektre.
figur 6 De simulerede spektre kan gemmes (tryk Save) og hentes igen (tryk Load).
CNMR-viewer. Fremvisning af beregnede protondekoblede C NMR-spektreVælg Spectrum | Open… . I dialogboksen "Load spectrum" åbnes csp-undermappen i mappen work2. Man kan nu vælge det stof fra databogen, hvis spektrum man ønsker at se. Fx er butanon stof nr. 29 på side 118 i databogen, dvs. C NMR-spektret er 118_29.csp. Programmet viser nu det beregnede C NMR-spektrum med en spektrometerfrekvens på 40 MHz i fuld skala fra –5 til 220 ppm. Man kan vælge en ny spektrometerfrekvens inden for området 10-1000 MHz. Vælg frekvensen 10 MHz og klik et vilkårligt sted i spektret. Spektret omregnes nu svarende til den nye spektrometerfrekvens.
figur 7
Da den relative hyppighed af 13C isotopen kun er 1.1%, ses der ingen kobling mellem nabostillede C-atomer. Derimod er der en kraftig kobling mellem nabostillede 13C og 1H atomer, (koblingskonstanten J er på 100-250 Hz, altså af samme størrelsesorden som forskellen i kemisk skift for de forskellige 13C atomer). For at gøre spektret enkelt at fortolke, vælger man derfor at dekoble til alle 1H atomer ved at anvende dobbelt resonans. Herved opnår man, at toppene for de enkelte 13C atomer optræder som singlets. Toppen ved 0.00 ppm er TMS signalet. I værktøjslinjen kan man vælge at få vist integralkurven, men som det fremgår af spektret, er intensitetsfordelingen ikke ligefrem proportional med antallet af C-atomer, der er repræsenteret ved de forskellige toppe. Kvaternære C-atomer og C-atomer, der ikke er bundet til H-atomer, vil oftest optræde med væsentligt lavere intensitet end de tilsvarende primære, sekundære og tertiære C-atomer. Man kan derfor ikke anvende integralkurven til protondekoblede C NMR-spektre. Der kan skelnes mellem de forskellige typer C-atomer ved at vælge de forskellige C-subspektre (Tools | C-subspectra…) .
Simuleret spektrometerkørselCNMR-vieweren kan give brugeren en illusion af at operere med et egentligt spektrometer, idet programmet giver mulighed for at vælge mellem forskellige måder at optage et 13C-spektrum på. Udover det traditionelle dekoblede 13C-spektrum, kan fx Off-resonans eller J-modulation give spektre, der bidrager med yderligere information i identifikationsarbejdet.
Off-resonans er en teknik, hvor 13C-1H koblingen reduceres til en brøkdel af dens faktiske værdi, og generelt fjernes langtrækkende kobling. I CNMR-vieweren bliver 13C-1H koblingen reduceret til 16 Hz. Herved begrænses omfanget af overlap mellem de forskellige opslittede toppe, og fortolkning af spektret bliver meget nemmere. Udfra koblingsmønsteret kan man let afgøre, om de forskellige C-atomer er primære, sekundære, tertiære eller kvaternære.
figur 8 Hvis koblingsmønsteret ikke ses tilstrækkeligt tydeligt, kan man som beskrevet i afsnittet om HNMR-vieweren ændre på spektrometerfrekvensen eller zoome ind på udvalgte områder af spektret.
J-modulation er en teknik, hvor toppe, der hidrører fra CH3- og CH-grupper peger nedad, mens toppe fra CH2- og C-grupper peger opad.
figur 9 OpløsningsmidletI eksperimentelle 13C-spektre vil opløsningsmidler, der indeholder C-atomer, give anledning til toppe i 13C-spektret. For hurtigt at kunne afgøre hvilke toppe i det eksperimentelle spektrum, der skyldes opløsningsmidlet, kan man i CNMR-vieweren specificere opløsningsmidlet, dets volumen og stoffets masse. Ved klik et vilkårligt sted i spektret eller ved tryk på Enter vises det genberegnede spektrum inklusiv opløsningsmidlets toppe med en intensitetsfordeling i overensstemmelse med den valgte stofkoncentration. CNMR-vieweren korrigerer ikke for opløsningsmidlets indflydelse på de kemiske skift i stoffet.
figur 10 Angivelse af data:Ved at trykke på Show kan man vælge at få vist diverse data i spektret: Chemical Shifts, Carbon Numbering, Integral Curve, Confidence Limits, Structure Window og Table of Chemical Shifts.
figur 11
gNMRgNMR er et program til simulering af NMR spektre udgivet af Cherwell Scientific Publishing. En demoudgave af programmet kan hentes fra: http://www.cherwell.com/gnmr/. Demoudgaven er en kopi af gNMR med nogle få undtagelser: det er ikke muligt at udskrive, gemme eller kopiere data til klippebordet. Denne gennemgang dækker den almindeligste anvendelse af gNMR og er tænkt som en hjælp til begyndere, der skal i gang med at bruge programmet. Det er ikke en fuldstændig gennemgang af programmet med alle dets muligheder, i så fald henvises der til den medfølgende on-line tutorial og hjælpefilerne samt evt. manualen, der følger med ved køb af programmet. Vi vil i det følgende simulere spektret for methansyreethylester HCOOCH2CH3. I figur 12 ses et spektrum af methansyreethylester, som er optaget med et Varian EM360 NMR-spektrometer.
figur 12 Nye data kan indsættes i gNMR på to måder for at kunne foretage en simulation: Simulation uden strukturformel, hvor man indsætter data om molekylet direkte i en tabel. Simulation med strukturformel, hvor man importerer en strukturformel fra et kemisk tegneprogram, fx ChemSketch 3.5
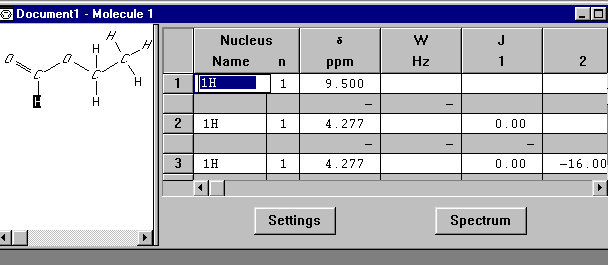
Simulation uden strukturformelData om methansyreethylester-molekylet skal indtastes. Efter at gNMR-programmet er startet, fremkommer et Welcome Window, her klikkes Cancel. Derefter vælges File|New , og et tomt "Molecule Window" fremkommer. "Molecule Window" ligner et regneark; det har kolonner til indtastning at kerner, skift, koblingskonstanter, etc. Første kolonne indeholder navnet på den kerne, 1H, som skal benyttes – den står der allerede. Flyt til næste kolonne med piletasterne. Her indtastes antal ækvivalente kerner. I tredje kolonne indtastes det kemiske skift. Værdierne fås fra en tabel over kemiske skift, eller de kan som her hentes fra det eksperimentelle spektrum. Fjerde kolonne kan indeholde individuelle liniebredder. Denne kolonne springes over, da der anvendes samme liniebredde i hele spektret. Resten af Molecule Window er en matrix med koblingskonstanter. For eksempel skal feltet i række 3 og kolonne 2 indeholde J23 , koblingskonstanten mellem kerne 2 og 3. Værdierne fås fra en tabel over koblingskonstanter. Når tabellen er udfyldt, skal den se ud som vist i figur 13.
figur 13 Spektrometerets frekvens skal ændres, da frekvensen for gNMR er forvalgt til 100 MHz, men spektret i figur 12 er optaget med et 60 MHz spektrometer. Vælg derfor Settings|File. I dialogboxen sættes frekvensen nu til 60 MHz. Klik OK. Spektret kan nu ses, klik Spectrum. I Spectrum Window fremkommer nu det beregnede spektrum af methansyreethylester. Integralkurven kan tilføjes på følgende måde: Klik med højre museknap på Spectrum window, vælg Integrate. Spektret skulle nu se ud som vist i figur 14.
figur 14 Bemærk overensstemmelsen med det eksperimentelle spektrum i figur 12. Det er en god øvelse at prøve at ændre på forskellige parametre (kemisk skift, koblingskonstanter og spektrometerfrekvens) og se hvilken virkning, det har på spektret. Man kan ændre i kemisk skift og koblingskonstanterne interaktivt på følgende måde: Vælg Molecule Window, anbring markøren i det felt, der ønskes ændret, klik Edit| Interactive xx. Der fremkommer en dialogbox, der gør det muligt interaktivt at ændre parameterens værdi og straks se virkningen i spektret (hvis der er plads til både Molecule Window og Spectrum Window på skærmen). Liniebredden kan ændres interaktivt ved at klikke med højre museknap på Spectrum Window, vælg Interactive Width. Simulation med strukturformelSom en alternativ metode til den ovenstående kan gNMR bruge et molekyles strukturformel til beregning af spektroskopiske parametre . Molekylet tegnes i et andet program, fx ChemSketch 3.5. Spektret for methansyreethylester skal igen simuleres. Det antages, at strukturformlen for molekylet er tegnet i ChemSketch 3.5 og eksporteret som en MDL-MOL fil. I gNMR importeres filen, vælg File|Import|MDL-MOL. I fil-dialogvinduet vælges MOL-filen. Derefter fremkommer Import Struktur vinduet; der ønskes ikke noget specielt, så klik OK. gNMR vil prøve at bestemme kemisk skift og koblingskonstanter ved brug af en database med kemisk skift samt simple additionsregler. Når programmet er færdig fremkommer Molecule Window, som vist i figur 15.
figur 15 Man skal betragte resultatet som en første tilnærmelse, idet der kan være nogen afvigelse, men metoden er et nyttigt udgangspunkt til produktion af data, som let kan ændres. Klik Spektrum. Uden at der er foretaget ændring i data, fremkommer spektret, som vist i figur 16. Der er her nogen afvigelse i kemisk skift, men erfaringen viser, at gNMR bestemmer rimelige data ud fra strukturformlen. Foretag evt. ændringer i kemisk skift og klik Recalculate.
figur 16 ExchangereaktionerI figur 17 ses ethanolspektret beregnet af gNMR på grundlag af en strukturformel tegnet i ChemSketch 3.5. Som man kan se, er der kobling mellem protonen i OH-gruppen og protonerne i CH2-gruppen, derfor fås multipletterne, som vist i figur 17.
figur 17 For at kunne forklare hvorfor protonen i hydroxygruppen normalt ses som en singlet, er man nødt til at tage hensyn til exchangereaktioner. Under normale forhold vil der være tilstrækkelig med sure urenheder til stede i opløsningen til at katalysere hurtig udskiftning af hydroxyprotonen. Protonen er ikke ved oxygenatomet længe nok til at koble med protonerne i CH2-gruppen. gNMR kan tage hensyn til intermolekylære exchangereaktioner på følgende måde. Importer først et ethanolmolekyle, Filer|Import. Find filen i fil-dialogvinduet. Vælg Molecule|New|Molecule2. Derefter importeres endnu et ethanolmolekyle på samme måde som det første. Vælg Molecule|Exchange for at åbne Exchange Window. De to strukturer er vist til venstre i figur 18, til højre skal exchangedata indtastes. Læg mærke til at protonerne er nummereret på følgende måde: 1-1 betyder proton 1 på molekyle 1; 2-3 betyder proton 3 på molekyle 2. En hurtig måde at indtaste exchangedata på er at klikke på protonen i OH-gruppen og derefter, med shift-tasten holdt nede, klikke på protonen i OH-gruppen i det andet molekyle. Skriv + for at fuldføre permutationen. Indtast 300 i Rate boksen og tryk Return. Indholdet i boksen nedenunder skifter til Fixed, som viser, at de nødvendige data er indtastet. Exchange Window vil derefter se ud, som vist i figur 18.
figur 18 Tryk Spectrum eller Recalculate for at se virkningen af exchangereaktionen. Derefter fremkommer spektret, som vist i figur 19. Foretag evt. ændring af rate for at se virkningen. Beregningerne kan godt tage lang tid afhængig af computeren.
figur 19
Organic Chemistry OnlineOrganic Chemistry Online CD-ROM with Workbook af Paul R. Young, University of Illinois at Chicago. Arbejdsbogen er på 206 sider. Published by Brooks/Cole, 1999. ISBN 0-534-35787-3. Kan bestilles online: www.thomson.com Pris: US $ 21.95 (ult. 1998). CD-ROM og arbejdsbog indeholder uafhængige moduler om 17 forskellige emner inden for organisk kemi. Et af modulerne handler om spektroskopi (IR, 1H NMR, 13C NMR, og MS) og giver mulighed for at arbejde interaktivt med spektroskopiske problemstillinger. Der er selvstændige forløb inden for hver af de spektroskopiske metoder, men der er også forløb, hvor de 4 forskellige spektroskopiske metoder integreres. Spektroskopiafsnittet afsluttes med en quiz. Et udvalg af disse forløb er tilgængelig på webadressen:
http://homework.chem.uic.edu/next.htm Klip fra Online-udgaven (CD-ROM) vises på figur 20 og figur 21.
figur 20 Spectroscopy med Review Sections, Problem Sets og Multiple Choice Quiz
figur 21 En opgave giver en bruttoformel samt mulighed for at se forskellige spektre for stoffet. Yderligere oplysninger om stoffet kan fås fra Analysis (se figur 21).
figur 22 I figur 22 ses billedet efter tryk på knappen Infrared. Hjælp, der vises i det nederste felt, kan fås ved at klikke på en top eller på Interpret, som der er klikket på her.
Infrared Spectroscopy Tutorial and Reference (IR Tutor)Infrared Spectroscopy Tutorial and Reference er et undervisningsprogram i IR-spektroskopi. Programmet er lavet af Charles B. Abrams, Colombia University i samarbejde med PERKIN ELMER. På adressen http://www.columbia.edu/cu/chemistry/edison/IRTutor.html findes yderligere oplysninger om programmet samt mulighed for at hente en demo af programmet. Programmet er delt op i 3 hovedafsnit: Introduction, Theory of Spectroscopy og Interpretation of Spectra. Hvert hovedafsnit er igen inddelt i et antal underafsnit, hvor man kan klikke sig frem gennem afsnittets sider. I Introduction gives en oversigt over de grundlæggende principper i spektroskopi, herunder en gennemgang af IR-spektrometret, både et klassisk instrument og et moderne (Fourier Transform). I Theory of Spectroscopy omtales, hvordan lys absorberes af molekyler. Både den klassiske model af et toatomigt molekyle og den kvantemekaniske model beskrives. Desuden forklares mere komplicerede vibrationer af molekylerne: vand, carbondioxid og pentan.
Interpretation of Spectra er det centrale afsnit i programmet. IR-spektrene for 13 stoffer bliver gennemgået i detaljer. Sammenhæng mellem absorptionsliner i spektrene og molekylets vibrationer bliver vist på en meget illustrativ måde. Programmet gør i høj grad brug af interaktiv grafik og avancerede computeranimationer, og det er derfor velegnet til at give en forståelse af grundlæggende principper i IR-spektroskopi.
Proton NMR Basics and Reference (NMR Tutor)Proton NMR Basics er et undervisningsprogram i NMR-spektroskopi fra Journal of
Chemical Education Software. Programmet er lavet at C. S. Judd, J. D. Morrisett, M. V.
Chari og J. L. Browning Houston Community College System, Houston. Proton NMR Basics
er en multimedie-tutor, som findes på CD-ROM. NMR Tutor er inddelt i fire afsnit: Introduction, The Instrument Room, The Classroom og The Laboratory. I Introduction gives et eksempel på, hvordan man bruger NMR-spektroskopi til at bestemme et ukendt stof. I The Instrument Room gives dels en gennemgang af den generelle teori for NMR-spektroskopi og dels vises, hvordan man betjener instrumentet. Der er desuden en detaljeret gennemgang af instrumentets opbygning. Grafik og animationer bliver i høj grad brugt til at underbygge forklaringerne. I The Classroom forklares, hvilken sammenhæng der er mellem molekylets struktur og NMR-spektret ud fra 6 stoffer (propan-2-ol, ethansyremethylester, benzen, ethansyreethylester, propanon og propensyremethylester). Atomer, der er ansvarlige for resonanstoppe, bliver fremhævet i en 3-D molekylmodel samtidig med toppen. Der er mulighed for at rotere 3-D modellen. I The Laboratory gives der en introduktion til den forskningsmæssige anvendelse af NMR inden for biokemi og medicin. Der vises, hvordan NMR-spektroskopi bruges til at studere proteinenzymet ribonuclease. Programmet giver en illustrativ indføring i NMR-spektroskopi og er et udmærket supplement til almindelige tekstbøger. Den digitale video, der viser betjeningen af NMR-spektrometeret, kompenserer i nogen grad for manglen på et rigtigt instrument. Udklipning af spektre og anden grafik med Screen Rip32Spektre, molekylformler og anden grafik kan overføres til et tekstbehandlingsprogram med programmet Screen Rip32. Programmet kan hentes som freeware: http://members.aol.com/progency/index.html.
Nu forsvinder Screen Rip32 vinduet automatisk, og næste gang der trykkes på musen, starter udklipningen. Vinduet, der skal klippes fra, skal derfor være aktivt. Hvis det ikke er aktivt, så gør det aktivt ved at klikke i det med musen.
Web-baserede opgaverSmå interaktive opgaver i tolkning af H NMR-spektreI dette afsnit vil vi kort introducere proceduren for arbejdet med små opgaver i tolkning af simple H NMR-spektre. Opgaverne ligger på en webside, hvor eleverne direkte og interaktivt kan arbejde med tolkningen. Opgaverne kan anvendes i forbindelse med et undervisningsforløb om spektroskopiske metoder. Forudsætningerne for at løse opgaverne er, at eleverne har fået gennemgået den grundlæggende teori inden for H NMR-spektroskopi, og at de er blevet fortrolige med tolkning af simple spektre. Det vil sige, at eleverne skal være bekendt med betydningen af det kemiske skift og baggrunden for toppenes opsplitning i multipletter. Herudover skal eleverne være i besiddelse af tabelværdier for kemiske skift (og eventuelt koblingskonstanter), fx fra Databog fysik kemi eller fra en tabel der ligger på websiden. Opgaverne er konstrueret, så de kan håndteres direkte fra en internetbrowser (Internet Explorer eller Netscape), og de er placeret på følgende adresser: http://www.ke.gymfag.dk/ikt/spektro1/ http://www.ke.gymfag.dk/ikt/spektro2/
Har man startet sin browser og skrevet ovennævnte adresse i adressefeltet, dukker følgende startbillede op:
figur 23 Herfra kan man klikke sig ind på de otte øvelsesopgaver. Det anbefales at starte med de lavest nummererede opgaver. Fra opgaverne er der genveje tilbage til siden med opgaveoversigten. Når en opgave er valgt, bliver der vist et skærmbillede af følgende type:
figur 24
Man har i dette billede mulighed for at få hjælp til tolkningen af H NMR-spektret ved at placere markøren på en af toppene og klikke med venstre museknap. Efter denne procedure dukker en lille hjælpetekst op i sidens nederste vindue.
figur 25 Det er tanken, at eleverne ved selvstændigt arbejde skal forsøge at tolke spektrene. Det betyder, at de, med baggrund i stoffets strukturformel og de informationer spektrene giver, skal foreslå en strukturformel for det pågældende stof. Den tabel med værdier for chemical shifts (kemiske skift) - som findes på websiden - dækker stort set behovet for information til løsning af opgaverne.
Når eleverne har et bud på en strukturformel, har man følgende muligheder for at kontrollere resultatet:— Ved at se om stoffet er med i den samling af NMR-spektre, der ligger på kemis hjemmeside. På startsiden er der links til kemis hjemmeside, hvorfra man kan hente H NMR-spektre (samt C NMR-spektre) for en lang række af stoffer i Databog fysik kemi. H NMR-spektrene kan kun ses i programmet HNMR-viewer 3.0, der kan hentes hjem fra ACDs hjemmeside (se ovenfor). — Ved at tegne molekylet i ChemSketch 3.5, exportere det som en mol-fil til simulatoren gNMR, hvor NMR-spektret så beregnes. En vejledning i brugen af ChemSketch 3.5 findes i et andet kapitel i dette hæfte, og en instruktion i brugen af gNMR findes i teksten ovenfor. — Ved at tegne molekylet i ChemSketch 3.5 og sende det til ACD beregner af NMR-spektre. (Det koster dog penge at få dette arbejde udført.) På websiden er der desuden reference til en samling på ca. tyve opgaver, hvor der både er H NMR- og IR-spektrum for det pågældende stof. Disse opgaver er beskrevet i det efterfølgende afsnit. Identifikation af organiske forbindelser ud fra molekylformel, IR- og NMR-spektrumVed identifikation af organiske stoffer ved hjælp af spektroskopi bruger man ofte en kombination af forskellige former for spektre. I denne opgave, som findes på kemis hjemmeside, skal man bestemme stoffets strukturformel, når man kender dets molekylformel, IR-spektrum og H NMR-spektrum. Stofferne er anbragt i en tabel, se figur 26. Der er i øjeblikket 18 stoffer. Opgaver mærket med "L" er forholdsvis lette opgaver. Henvisning til opgaverne findes på side *.
figur 26 Første søjle angiver nummeret på stoffet, og anden søjle angiver stoffets molekylformel. Ved at klikke på feltet i tredje søjle ses stoffets IR-spektrum, og ved at klikke på feltet i fjerde søjle ses dets H-NMR-spektrum. Denne tekst fremkommer ved at kikke på Information om opgaverne. IR-spektrene er fremstillet med en IR-simulator for instrumentet Perkin Elmer Model
1310 IR-spektrometer. Analyserne er i væskefase i natriumchloridkrystal. NMR-spektrene er
fremstillet med en NMR-Simulator for Varian EM360 NMR-spektrometer ved frekvensen
60 MHz. Simulatorerne er lavet af Paul Schatz, University of Wisconsin, Madison og
forhandles af firmaet Falcon: Tolkningen af spektrene er i nogen grad gjort interaktiv ved, at der er indlagt "hot-spots" på spektrene, dvs. at man kan klikke på visse dele af spektrene og få oplysninger som hjælp til opklaring af stoffernes struktur. Hvis man klikker på IR-spektret et vilkårligt sted, får man oplysning om de strukturer, der giver anledning til absorption i det pågældende område (området har en bredde på ca. 200 cm-1). Foruden oplysning om struktur angives det nøjagtige absorptionsinterval i cm-1, intensitet og evt. form. På NMR-spektrene kan man klikke på de resonanstoppe, der er i spektret. Man får da oplysning om strukturer, der giver resonans ved toppens kemiske skift eller tæt ved (ca. 0,5 ppm). Der er mulighed for at veksle mellem IR-spektret og NMR-spektret, ligesom man kan vende tilbage til startsiden med opgaverne. De oplysninger, man kan få interaktivt, svarer nogenlunde til oplysningerne i Databog fysik kemi. I nogle tilfælde kan det være nødvendigt at søge oplysninger i større tabelværker, sådanne findes på kemis hjemmeside. Et forslag til strukturformel kan kontrolleres ved at sammenligne det ukendte stofs spektrum med spektre af kendte stoffer. Det kan gøres på følgende måde: — H NMR-spektret kan man sammenligne med et simuleret spektrum af forslaget til strukturformel. Strukturformlen kan tegnes i ChemSketch 3.5 og eksporteres som MOL-fil. Derefter kan filen importeres i NMR-simulatoren gNMR eller sendes til ACD beregner af NMR-spektre. Man kan også benytte den samling af spektre, der ligger på kemis hjemmeside. — IR-spektre kan hentes fra http://webbook.nist.gov/chemistry/form-ser.htm.
Spektroskopiopgaver, der lægger op til brug af computerHer følger 3 opgaveeksempler. Opgaverne kan løses ved hjælp af tabeller i Databog fysik kemi samt tabeller placeret på kemis hjemmeside. Endvidere anvendes programmer, som omtalt i teksten ovenover.
Opgave 1Et organisk stof analyseres ved en elementaranalyse. Stoffet viser sig udelukkende at indeholde C, H og N. Ved fuldstændig forbrænding af 0,7560 g af stoffet blev der dannet 1,6880 g CO2 og 1,0368 g H2O. a. Beregn stoffets empiriske formel. Ved massespektroskopi bestemmes stoffets molare masse til 59,13 g/mol. b. Bestem stoffets molekylformel. c. Tegn de mulige strukturformler af stoffet. Nedenstående figur 1 viser stoffets 1H NMR-spektrum.
figur 1. 1H-NMR-spektret af stoffet. Aksen angiver kemisk skift i ppm. Integralkurver er indtegnet. d. Bestem forholdet mellem antallet af H-atomer i de forskellige grupper af kemisk ækvivalente H-atomer og kommenter beliggenheden af disse. e. Forklar koblingsmønsteret i NMR-spektret og identificer stoffet. f. Tegn stoffets strukturformel i ChemSketch og beregn stoffets H NMR-spektrum i gNMR programmet.
Opgave 2
Stoffet but-1-en reagerer med hydrogenchlorid, og danner to stoffer A og B. De to produkter adskilles, og der optages 1H NMR-spektre af A og B.
figur 1. 1H-NMR-spektrum af stof A. Aksen angiver de kemiske skift i ppm. Integralkurver er indtegnet.
figur 2. 1H-NMR-spektrum af stof B. Aksen angiver de kemiske skift i ppm. Integralkurver er indtegnet. a. Identificer A og B idet du inddrager integralkurver, kemiske skift og koblingsmønstre i de to 1H NMR-spektre i din argumentation. b. Kontroller dit svar ved at tegne A og B i ChemSketch og få beregnet 1H NMR-spektrene i gNMR programmet. c. Gør rede for hvilket af de to stoffer du forventer, der dannes mest af.
Opgave 3Et organisk stof indeholder kun C, H og O. Ved massespektroskopi bestemmes stoffets molare masse til 58,09 g/mol. Stoffets kemiske egenskaber:
I figur 1 vises det protondekoblede 13C NMR-spektrum af stoffet.
figur 1. Det protondekoblede 13C NMR-spektrum af stoffet. De kemiske skift er angivet i ppm. Signalet ved 0 ppm skyldes TMS. a. Anfør antallet af C-, H- og O-atomer i molekylet. I figur 2 vises off-resonans 13C NMR-spektret af stoffet.
figur 2. Off-resonans 13C NMR-spektret af stoffet. De kemiske skift er angivet i ppm. Signalet ved 0 ppm skyldes TMS. b. Gør rede for hvor mange H-atomer de forskellige C-atomer er bundet til. c. Gør rede for C-atomernes hybridisering. d. Gør rede for hvilken funktionel gruppe stoffet indeholder. e. Tegn stoffets strukturformel. f. Brug HNMR-vieweren til at vise 1H NMR-spektret af stoffet. g. Forklar det komplicerede koblingsmønster i stoffets 1H NMR-spektrum.
Spektroskopiopgaver, som kan løses ved hjælp af Databog fysik kemiHer følger 3 opgaveeksempler. Flere opgaver af denne type ligger på kemis hjemmeside – se side *.
Opgave 1Et organisk stof indeholder i masseprocent 66,7% C og 11,1% H. Stoffet har følgende IR- og 1H NMR-spektre:
figur 1. IR-spektrum.
figur 2. NMR-spektrum. Kemisk skift i ppm. Integraler er indtegnet. Ifølge stoffets massespektrum er den molare masse 72 g/mol. a. Opskriv stoffets molekylformel. b. Forklar ud fra IR-spektret, hvilke vibrationer der er i stoffet. c. Forklar hvilke oplysninger 1H NMR-spektret giver om stoffet. Det vides endvidere, at stoffet har et kogepunkt på 80 ° C, samt at det ikke kan oxideres af Tollens reagens. d. Opskriv stoffets strukturformel og navn. Opgave 2.Nedenfor vises IR- og 1H NMR-spektret af det organiske stof CaHbOc.
figur 1. IR-spektrum.
figur 2. NMR-spektrum. Kemisk skift i ppm. Integraler er indtegnet. a. Beskriv hvilke oplysninger IR-spektret giver om molekylet. Opskriv herunder både en funktionel gruppe, som molekylet indeholder, samt en det ikke indeholder. b. Ved fuldstændig forbrænding af 1,503 g stof fås 3,301g CO2 og 1,801g H2O. c. Stoffet kan oxideres af Cr2O72- i sur opløsning,
idet der dannes Cr3+-ioner samt et aldehyd. d. Opskriv og afstem reaktionsskemaet for den omtalte redoxreaktion. e. Forklar 1H NMR-spektrets udseende. Opgave 3.Isomere butanoler. a. Opskriv strukturformler og systematiske navne for de 4 isomere butanoler. Nedenfor vises IR-spektret for et af de isomere molekyler, samt 1H NMR-spektre for 2 andre af de isomere butanoler. b. Forklar IR-spektrets udseende. c. Hvilken af de 4 isomere vil have det simpleste NMR-spektrum? d. Forklar de 2 viste NMR-spektre idet kemisk skift, koblingsmønster og integraler
omtales.
figur 1. IR-spektrum.
figur 2. NMR-spektrum. Kemisk skift i ppm. Integraler er indtegnet.
figur 3. NMR-spektrum. Kemisk skift i ppm. Integraler er indtegnet. EksamenEksempel på eksamensspørgsmål i NMR-spektroskopi i tilknytning til kapitlet: Forudsætninger:I forberedelseslokalet har eleven adgang til de sædvanlige hjælpemidler. Herudover er der adgang til en computer, der er forsynet med følgende programmer: HNMR-viewer, CNMR-viewer, ChemSketch 3.5 og gNMR. I tilfælde af at det ikke lykkes for eleven at finde frem til strukturen for molekylet, ligger der filer på computeren (under anonymiserede navne, som kun er kendt af læreren). Filerne kan bruges i fortsættelsen med simuleringen i gNMR. 1H-NMR-spektroskopiPå vedlagte figur 1 findes 1H-NMR-spektret for et stof med molekylformlen C5H10O2 og det systematiske navn ethylpropanoat. Der ønskes en gennemgang af de overvejelser, du skal gøre for at forklare stoffets strukturformel ud fra spektret. Du skal herunder forklare begreberne integralkurve, kemisk skift og finstrukturopsplitning. Under eksaminationen kan du demonstrere en simulation af et NMR-spektrum for det pågældende stof med programmet gNMR. (Husk, at du først er nødt til at tegne strukturformlen i ChemSketch 3.5 og importere strukturen i gNMR-programmet som en mol-fil.)
Figur 1: 1H-NMR-spektrum for ethylpropanoat |
![[ Billede: Undervisningsministeriets logo ]](http://www.uvm.dk/images/uvmlogo.gif)
![[ Billede: skærmbillede med graf ]](212.gif)
![[ Billede: skærmbillede med graf ]](214.gif)
![[ Billede: skærmbillede med graf ]](215.gif)
![[ Billede: Dialogboks ]](217.gif) figur 4
figur 4![[ Billede: skærmbillede med graf ]](218.gif)
![[ Billede: Dialogboks ]](219.gif)
![[ Billede: skærmbillede med graf ]](220.gif)
![[ Billede: skærmbillede med graf ]](221.gif)
![[ Billede: skærmbillede med graf ]](222.gif)
![[ Billede: skærmbillede med graf ]](223.gif)
![[ Billede: skærmbillede med graf ]](224.gif)
![[ Billede: skærmbillede med graf ]](225.gif)
![[ Billede: Dialog boks ]](226.gif)
![[ Billede: skærmbillede med graf ]](227.gif)
![[ Billede: skærmbillede med graf ]](229.gif)
![[ Billede: skærmbillede med graf ]](230.gif)
![[ Billede: Dialogboks ]](231.gif)
![[ Billede: skærmbillede med graf ]](232.gif)
![[ Billede: skærmbillede fra Organic Chemistry's hjemmeside ]](233.gif)
![[ Billede: skærmbillede fra Organic Chemistry's hjemmeside ]](234.gif)
![[ Billede: skærmbillede fra Organic Chemistry's hjemmeside ]](235.gif)
![[ Billede: skærmbillede fra opgaveside ]](236.gif)
![[ Billede: skærmbillede af opgave set fra ens egen browser ]](237.gif)
![[ Billede: skærmbillede af opgave set fra ens egen browser ]](238.gif)
![[ Billede: skærmbillede fra informationsside om opgaverne ]](239.gif)
![[ Billede: skærmbillede med graf ]](240.gif)
![[ Billede: tegning af et molekyles opbygning ]](241.gif)
![[ Billede: skærmbillede med graf ]](242.gif)
![[ Billede: skærmbillede med graf ]](243.gif)
![[ Billede: skærmbillede med graf ]](244.gif)
![[ Billede: skærmbillede med graf ]](245.gif)
![[ Billede: skærmbillede med graf ]](246.gif)
![[ Billede: skærmbillede med graf ]](247.gif)
![[ Billede: skærmbillede med graf ]](248.gif)
![[ Billede: skærmbillede med graf ]](249.gif)
![[ Billede: skærmbillede med graf ]](250.gif)
![[ Billede: skærmbillede med graf ]](251.gif)
![[ Billede: skærmbillede med graf ]](252.gif)
![[ Billede: skærmbillede med graf ]](253.gif)